Help
[1] Overview
[2]
Browse
[3] Search
[4] High-throughput
[5] Drug Discovery
[6] Download
[7]
Contact
[1] Overview
Protein-coding RNAs refer to RNAs that
encode proteins. This kind of RNAs control various biological
functions to maintain morphology and functions of tissues, whose
dysfunction often influences cancer development. Accumulating
researches have proven that cancers related drugs can target
protein-coding RNAs. In addition, protein-coding RNAs may play
critical roles in drug resistance. These researches produced
substantial data such as literatures and high-throughput microarray
which were hard to be screened out by researchers. To collect and
annotate these data, we developed DREAM database (Figure 1),
a comprehensive manually curated database including protein-coding
RNAs and drug targets or drug sensitivity associations. Current
version contains 1601 scientific literatures and 195
high-throughput microarray data. Each entry in DREAM database
contains detailed information on RNAs, drug, cancer, and other
information.
The overview of the database is as
followings:

Figure 1. The overview of the
DREAM database.
[2] Browse
To browse protein-coding RNAs and cancer
drugs association data in the database, please click the menu
"Browse". All the data were divided into two parts called ‘Drug
intervention’ and ‘Drug resistance’. Users can browse all entries
in three ways: by compound name, by gene name, or by disease name.
In ‘Drug intervention’ part, take a browse "glioma"-related
protein-coding RNAs and drugs as an example. To browse the entries
for glioma, please click "Disease name" and select the "glioma"
option. The browse result will be displayed in the right panel as
presented in Figure 2A. In ‘Drug resistance’ part, take a
browse "cisplatin"-related protein-coding RNAs and cancers as an
example. To browse the entries for cisplatin, please click
"Compound name" and select the "cisplatin" option. The browse
result will be displayed in the right panel as presented in Figure
2B.

Figure 2. The browse interface of the DREAM
database.
[3] Search
DREAM database provides a fuzzy search
function. To search data in the database, please click the menu
"Search". Users can search all entries in three ways: search by
compound, or/and search by feature, or/and search by disease. The
‘Search’ page is displayed in Figure 3.
1. choose
drug intervention search or drug resistance search;
2.
input the interested drugs, protein-coding RNAs, or disease for
search.
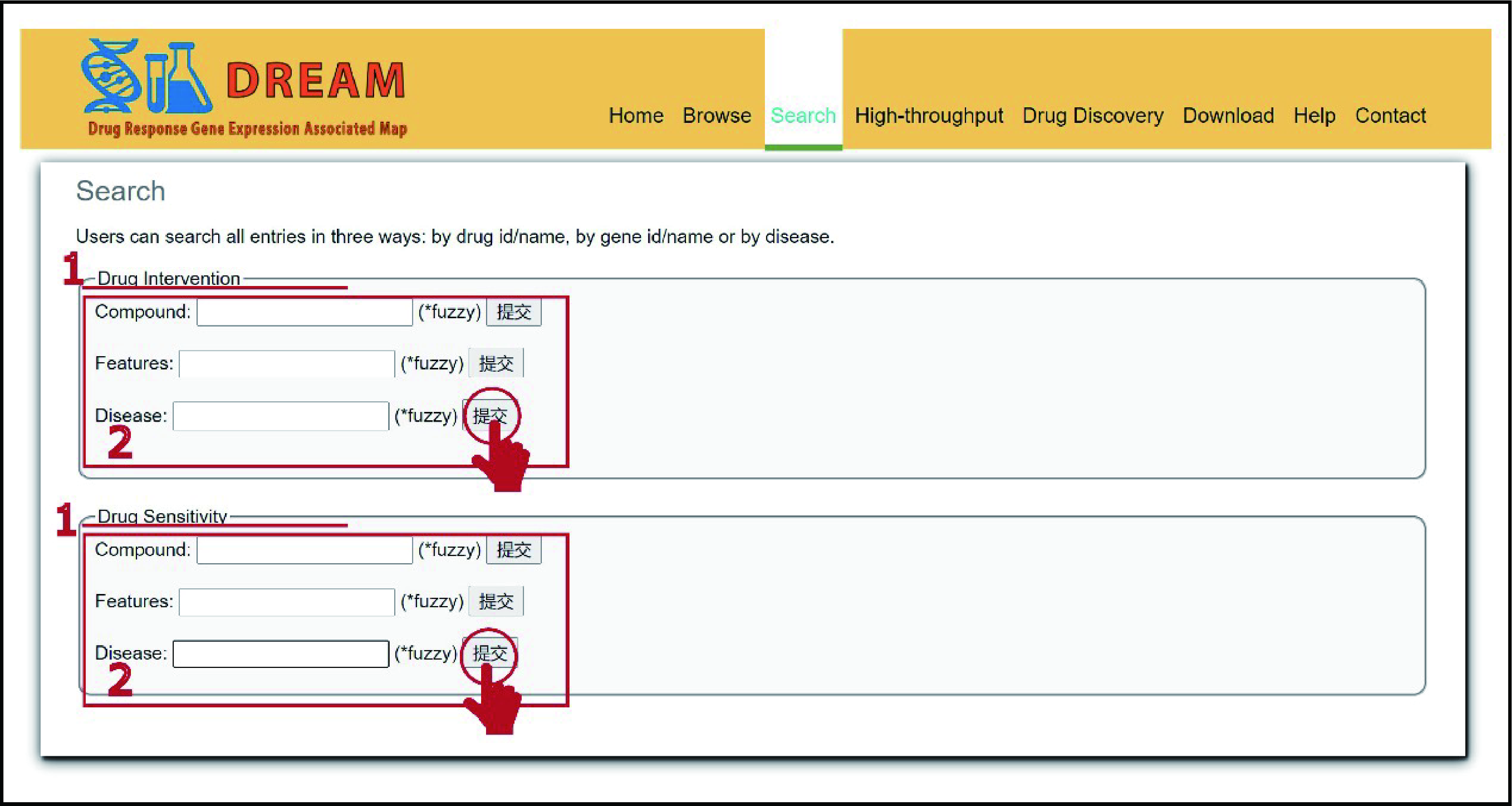
Figure 3. The search interface of the DREAM
database.
[4] High-throughput
The current version of DREAM
database contains 195 high-throughput microarray data across 36
cancer subtypes, including 94 protein-coding RNAs and drug
intervention associations microarray data and 101 protein-coding
RNAs and drug resistance associations microarray data. To browse
and search these data in our database, please click the menu
"High-throughput". We provide three search methods: search by
compound, search by feature, or search by disease. User can set the
statistical significance (p-values) and biological significance
(fold changes) for different high-throughput microarray data in
order to quickly identify the most interesting gene candidates
associated with drug targets or drug resistance.The p-values and
fold changes are calculated on the basis of case groups relative to
control groups. In “drug intervention” module, the case groups are
disease cells with drug intervention. The control groups are
disease cells with placebo intervention such as PBS, DMSO. In “drug
sensitivity” module, the case groups are drug resistant disease
cells and the control groups are normal disease cells.In addition,
we also offer users an interactive visualization tool such as
volcano plot and gene enrichment analysis such as GO annotations
and KEGG pathways analysis. The ‘High-throughput’ page is displayed
in Figure 4 taking ‘temozolomide’ as an example.
1. click to choose search by compound;
2. input user’s
interested drug ‘temozolomide’ and set special cut-off value such
as ‘fold change>2’ and ‘p value< 0.05’;
3. detailed
information returns in search results;
4. click to perform
function analysis.

Figure 4. The high-throughput
interface of the DREAM database.
[5] Drug Discovery
In ‘Drug Discovery’ page, user
can calculate the correlation coefficient between the drug's gene
expression signature and disease's expression signature to
repurpose and identify novel drug indications in different cancers.
The raw R code was displayed in the below table. The “drug's gene
expression signature” is extracted from drug-disease related gene
expression profiles in DREAM. The “disease's expression signature”
is gene sets with expressed protein-coding RNAs in disease groups
relative to healthy control groups. The “disease's expression
signature” demands user upload by themselves and must be composed
of gene symbol and fold changes.
## import user data
data <- read.table(<user_data>, header=T, sep = "\t", quote = "")
input.gene.list <- data[,1]
input.fd.list <- as.numeric(as.character(unlist(data[,2])))
input.dat <- data.frame(input.gene.list = input.gene.list, input.fd.list = input.fd.list)
rownames(input.dat) <- input.gene.list
## import reference data
dat <- read.table(<reference_data>, header = T, sep = "\t", quote = "")
dat$ID <- paste0(dat$Drug.id, "---", dat$Drug.name, "---", dat$Dataset.ID, "---", dat$Disease)
dat.ID.list <- unique(dat$ID)
result <- t(sapply(dat.ID.list,function(x){
#print(x)
drug.id <- strsplit(x,"---")[[1]][1]
drug.name <- strsplit(x,"---")[[1]][2]
dataset.id <- strsplit(x,"---")[[1]][3]
disease.id <- strsplit(x,"---")[[1]][4]
tmp.gene.list <- dat$Gene.id[dat$Dataset.ID==dataset.id&
dat$Drug.id==drug.id&
dat$Disease==disease.id]
tmp.fd.list <- dat$Foldchange[dat$Dataset.ID==dataset.id&
dat$Drug.id==drug.id&
dat$Disease==disease.id]
tmp.dat <- data.frame(tmp.gene.list=tmp.gene.list,
tmp.fd.list=as.numeric(as.character(unlist(tmp.fd.list))))
rownames(tmp.dat) <- tmp.gene.list
overlap.gene <- intersect(input.gene.list,tmp.gene.list)
cortest <- cor.test(input.dat[overlap.gene,"input.fd.list"],
tmp.dat[overlap.gene,"tmp.fd.list"])
c(drug.id,drug.name,dataset.id,disease.id,cortest$estimate[[1]],cortest$p.value)
}))
result <- data.frame(result)
colnames(result) <- c("drug_id","drug.name","dataset_id","disease","cor","p")
result$adj.p <- p.adjust(as.numeric(as.character(unlist(result$p))))
result.filter <- result[result$adj.p<0.05,]
result.filter$cor <- format(as.numeric(as.character(unlist(result.filter$cor))), scientific = TRUE,digit = 3)
result.filter$p <- format(as.numeric(as.character(unlist(result.filter$p))), scientific = TRUE, digit = 3)
result.filter$adj.p <- format(result.filter$adj.p, scientific = TRUE, digit = 3)
options(digits = 3)
write.table(result.filter, <output_file>, row.names = F, sep = "\t", quote = F)
In ‘Drug Discovery’ page, firstly user need to upload disease's
expression signature including gene symbols and fold change
(relative to a healthy control) by themselves. Then, our database
will match and calculate the correlation coefficients based on the
database containing drug's gene expression signature. After a
while, the database will output the results including correlation
coefficient, drugs, and cancers. The ‘Drug discovery’ page is
displayed in Figure 5.

Figure
5. The drug-discovery interface of the DREAM database.
[6] Download
To download data in DREAM database,
please click the menu ‘Download’. The DREAM provides two formats of
downloadable files in TEXT and Excel formats, respectively. The
‘Download’ page is displayed in Figure 6.

Figure
6. The download interface of the DREAM database.
[7] Contact
If you have any questions, suggestions
or comments, please contact us by e-mails. In ‘Contact’ page, there
are the details of two corresponding writers of our team.